CHAPTER 7. ONE-ELECTRON TRANSFER: RADICAL REACTIONS
(Arthur Cammers, Ashley Jolly Steelman, and Susan Odom, University of Kentucky, 2019; with excepts from Timothy Soderberg’s, “Organic Chemistry with a Biological Emphasis II,” 2016)
Learning Objectives:
1. Define the term radical.
2. Draw resonance structures for radical compounds. Identify the HOMO of simple radical molecules.
3. Identify the weakest carbon-hydrogen bond in a compound.
4. Recognize the common patterns that occur in radical mechanisms.
5. Determine the reagents used and products formed for radical halogenation reactions.
6. Draw the products for radical halogenation including relevant regiochemical and stereochemical considerations.
7. Draw the mechanism for radical halogenation including initiation, propagation, and termination steps.
8. Determine the reagents used and products formed for allylic bromination reactions.
9. Understand why allylic and benzylic bromination works when other organic substrates fail to brominate.
10. Draw the products for allylic bromination including relevant regiochemical and stereochemical considerations.
11. Draw the mechanism for allylic bromination including initiation, propagation, and termination steps.
12. Explain why a small amount of CFCs can do significant damage to the ozone layer.
13. Explain why it is a safety concern to store some sulfur and oxygen containing materials in the presence of oxygen gas.
14. Draw the products for radical hydrohalogenation including relevant regiochemical and stereochemical considerations.
15. Draw the mechanism for radical hydrohalogenation.
16. Describe the synthetic use of radical halogenation.
17. Identify the oxidation state on carbon atoms, and describe whether reactions involve carbon oxidation, reduction, or neither.
18. Draw the products for dissolving metal reduction including relevant regiochemical and stereochemical considerations.
19. Draw the mechanism for dissolving metal reduction including initiation, propagation, and termination steps. Which MOs are occupied?
20. Draw the products for radical polymerization including relevant regiochemical and stereochemical considerations.
7.1 INTRODUCTION TO RADICALS
(Arthur Cammers, University of
Kentucky, 2019)
 Image:
Magnet and sample mount of the electron paramagnetic resonance (EPR)
spectrometer at the University of Kentucky. EPR spectrometers are instruments used
to detect radicals.
Image:
Magnet and sample mount of the electron paramagnetic resonance (EPR)
spectrometer at the University of Kentucky. EPR spectrometers are instruments used
to detect radicals.
Think about putting two atoms together to make a molecule and the MO energy diagram that accompanies that process. When we think about bond strength—bond dissociation energy, we think about forming the bond from two atoms or molecules with odd electrons. Bond formation pairs the electrons. Since electrons are more stable in larger orbitals, and stabilized by two positive nuclei, this process decreases the energy of the atoms involved. The two electrons enjoy the increased freedom; they can avoid each other by distinguishing their quantum numbers at the spin level. For most molecules, the energy of the electrons decreases upon bond formation, but we can imagine a molecule that might have a high degree of π-bonding (conjugation) that would not stabilize upon bond formation.

Figure A. Examples of radical species: (a) tris(4-bromophenyl)ammoniumyl hexachloroantimonate, (b) 3-cyano-PROXYL, (c) galvinoxyl, and (d) α,γ-bisdiphenylene-β-phenylallyl.
These are a few ■ commercially available radicals. There are many other stable radicals in the literature. Again, the recipe is enough bonding conjugation and enough of a steric environment to render the localization of the odd electron in a bond higher in energy than it would be roaming around in a π-system. The names of the radicals above are difficult to parse into anything sensible, but you will notice something interesting, they all end in ‘yl’.
This chemical suffix ‘yl’ means radical. You will recall that we named substituents on molecules by their radical fragments. Notice below that the C atom and bound H atoms are explicit when the valence is open at the C atom. Drawing radicals this way will give you clarity when thinking about radical mechanisms and others reading your communications will find them less ambiguous. None of the radicals below can be bottled. They would dimerize.
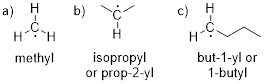

Exercise 7.1.1 Predict the reaction product if you could generate the methyl radical (a) in a reaction vessel. Do the same for structures b – f. Use curved arrows to show the mechanism of the reactions.
7.1.1 Radical Mechanisms
Previously we
noted that in heterolytic two-electron processes a double headed arrow is used
to indicate the movement of the two electrons. Radical reactions involve one
electron, usually neutral homolytic changes in bonding. In this case, as we
indicated previously, to show the reaction mechanism a single head on the arrow
is used.
There are some
notoriously weak bonds that can initiate radical processes; we’ll unpack that
statement just a little later. To show the thermal homolytic reactions of these
bonds you may do the following.
![]()
Often light initiates bond cleavage by molecular EM absorption and promotion to the electronic excited state. The ■ Frank Condon Principle states that vibronic excitation must immediately accompany electronic excitation. Many molecules with weak bonds have very short lifetimes as electronic exited states; they tend to dump this energy by breaking the bond. To show the photolysis of these bonds do the following; indicate the excited state with an asterisk.
![]()
Radical species can break σ bonds. When they take an H atom (•H) from other molecules this is referred to as H atom abstraction. It is NOT to be confused with proton (H+) transfer, which is a heterolytic process. Since radicals can break σ bonds it is no surprise that they can also break π bonds by adding directly to them; this process is called radical addition. See the examples below.
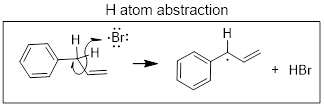
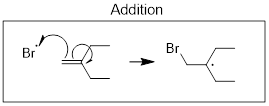
Spin is conserved in radical reactions just like charge is conserved. A radical, by definition, has an odd number of electrons. When two radicals react, the product is electron paired (even number of electrons). When a radical reacts with a regular electron-paired molecule the product has an odd-number of electrons; it is a radical.
7.2 RADICAL STABILITY
(Arthur Cammers, University of
Kentucky, 2019)
Notice in the addition reaction above that the Br atom did not add to the most substituted C atom. The reaction made the most stable radical out of two possibilities.
![]()
This mode of addition is not observed.
This mode of addition is not observed because this pathway and this product are higher in energy than the addition reaction that generates the more substituted product which comes from the more substituted radical. Let’s compare the 1-bromo-2-yl intermediate versus the 2-bromo-1-yl intermediate that is not generated by the addition. When a reaction selects one or more structural isomers as a product or selects one structurally unrelated product we say that the reaction is ■ regioselective. There are many such terms, and with the knowledge attained at this point you can likely correctly guess what enantioselectivity, diastereoselectivity and stereoselectivity mean. All of these ‘…ivities’ are mandated by thermodynamics, by the stabilities of electrons in transition states or ground states depending on the nature of the reaction. At least we know how to look for an explanation of the regioselectivity of the addition reaction. It is something we discussed previously.

The orbital containing the odd electron is mostly p in character and it can align partially with σ bonds but only with one at a time. These parallel interactions between the radical orbital and σ-σ* stabilize the electrons involved. In the less stable radical, I can show you only one parallel interaction with σCC. The more stable radical has three parallel interactions with σCH. Furthermore, a σCC‑p interaction is not as stabilizing as a σCH‑p interaction. This is because radicals as hextets are electron-poor and σCH is a better electron donor than σCC. The parallel interaction above is termed ■ hyperconjugation, a general term for the stabilizing interaction of σ bonds with unoccupied orbitals, π* or partially occupied orbitals. As with regular conjugation the more of it that there is the more stabilizing the interactions are.

The structures of radicals above with methyl less stable than primary (1°), which is less stable than secondary (2°) and the tertiary radical is the most stable (3°). However, 3° radicals are still reactive. In general, they cannot be bottled. Remember: You need real π bonding to bottle a radical, with real conjugation.
Exercise 7.2.1 Put the 2-propyl radical in the appropriate conformation for hyperconjugative interaction. Show this interaction in a ■ Newman Projection.
Exercise 7.2.2 We know that sp and sp2 C‑H bonds have higher BDEs than sp3 C-H bonds due to their higher percent s character. However, previously we treated all C‑H bond dissociation energies as the same. Now you know better. Which BDE is greatest, label that one 4, and the next one, 3 and so on. See the key below for ballpark estimations of the real values.

7.3 CHLORINATION OF ETHANE
(Timothy Soderberg, Organic Chemistry with a Biological Emphasis II, 2016, 349-351)
Because of
their high reactivity, free radicals have the potential to be extremely
powerful chemical tools - but as we will see in this chapter, they can also be
extremely harmful in a biological/environmental context. Key to understanding
many types of radical reactions is the idea of a radical chain reaction.
Radical chain
reactions have three distinct phases: initiation, propagation, and termination.
We'll use a well-known example, the halogenation of an alkane such as ethane,
to illustrate. The overall reaction is:
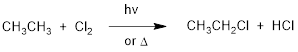
The initiation
phase in a radical chain reaction involves the homolytic cleavage of a weak
single bond in a non-radical compound, resulting in two radical species as
products. Often, heat or light provides the energy necessary to overcome an
energy barrier for this type of event. The initiation step in alkane
halogenation is homolysis of molecular chlorine (Cl2) into two
chlorine radicals. Keep in mind that virtually all radical species, chlorine
radicals included, are highly reactive.
![]()
The propagation
phase is the 'chain' part of chain reactions.

Once a reactive
free radical (chlorine radical in our example) is generated in the initiation
phase, it will react with relatively stable, non-radical compounds to form a
new radical species. In ethane
halogenation, a chlorine radical generated in the initiation step first reacts
with ethane in a hydrogen abstraction step, generating HCl and an ethyl radical
(part a above). Then, the ethyl radical
reacts with another (non-radical) Cl2 molecule, forming the
chloroethane product and regenerating a chlorine radical (part b above). This process repeats itself again and again,
as chlorine radicals formed in part (b) react with additional ethane molecules
as in part (a).
The termination
phase is a radical combination step, where two radical species happen to
collide and react with each other to form a non-radical product and 'break the
chain'. In our ethane chlorination
example, one possible termination event is the reaction of a chlorine radical
with an ethyl radical to form chloroethane.

Because radical
species are so reactive and short-lived, their concentration in the reaction
mixture at any given time is very low compared to the non-radical components
such as ethane and Cl2. Thus, many cycles of the chain typically
occur before a termination event takes place.
In other words, a single initiation event leads to the formation of many
product molecules.
(Ashley Jolly Steelman, 2019)
Let’s work
through a practice problem.
Example 7.3.1 Complete the mechanism for the radical chlorination of methane. The overall reaction is shown.

Solution:
Step 1: Initiation - we need to create our reactive radical species by light-induced homolysis.
![]()
Step 2: Propagation – the reactive radical can interact with our starting materials. There are two steps in the propagation phase of the reaction; we will call them (a) and (b).


Note: the reformation of a reactive chlorine radical in propagation (b) causes the propagation process to keep repeating until termination occurs.
Step 3: Termination – the coupling of any two radical species will stop the reaction.

Note: If large
amounts of Cl2 is present, polychlorination can occur where all
hydrogens of methane can be abstracted and replaced with chlorine atoms. [
Cammers: The concentration of Cl2 should be low, but also the
chemist should not run the reaction to completion. The longer the reaction runs
the more polychlorinated products will be obtained. In this case, if
chloromethane is the desired product. It can be readily separated from methane,
and other polychlorinated methane derivatives by distillation. ] Unless otherwise told, we will assume that the
conditions of the chlorination reaction are controlled to only produce the
monohalogenation product.

7.4 THERMODYNAMICS of HALOGENATION
(Arthur Cammers, University of
Kentucky, 2019)
Do other halogens
permit radical halogenation: F2, Br2, or I2?
Often just asking a question requires deeper thinking about the nature of the
phenomenon; this is one of those questions. In answering it, let us reach into
our kit bag for the reaction energy diagram and use it to characterize the
nature of radical reactions.

Whether it is heat or light initializing the reaction, the intermediates in the radical chain must take place in an energetic realm above the regular stabilities of the spin-paired ground states because a bond is always a broken in B, C or D.
CH3CH2CH3 + Cl2 ─hν ® CH3CHClCH3 + HCl
On the reaction energy diagram above for the chlorination of propane, A = CH3‑CH2‑CH3 + 3/2 Cl2; B = CH3‑CH2‑CH3 + Cl2 + Cl•; C = (CH3)2CH• + HCl + Cl2; D = (CH3)2CHCl + HCl + Cl•; E = (CH3)2CHCl + HCl + ½ Cl2. States B, C and D cycle Cl•, in the sea of reactants. This reaction energy diagram should remind you of the reaction energy diagram for catalyzed processes. From that perspective, you should be able to imagine some hypothetical, higher-energy process with fewer intermediates analogous to some ‘uncatalyzed’ reaction. The radical chain provides a stepwise path to products similar to catalytic intermediates in a ■ catalyzed process. ΔGrxn < 0 for a catalyzed reaction to proceed. Remember that catalysis changes the reaction rate, not the equilibrium. Since the net reaction is: CH3‑CH3 + X2 ® CH3‑CH2X + HX, we know how to quickly evaluate whether the reaction is thermodynamically favorable by calculating the enthalpy of the reaction using BDE.
|
BDE (kJ /mol) |
||||
|
H─CH(CH3) |
X |
X─X |
X─CH2CH3 |
H─X |
|
412.5* |
F |
157.0 |
462.8* |
570.1* |
|
|
Cl |
242.6 |
356.5* |
431.6* |
|
|
Br |
193.9 |
309.2* |
366.3* |
|
|
I |
152.4 |
238.5* |
298.4* |
* “Bond Dissociation Energies of Organic Molecules” S. J. Blanksby, G. B. Ellison Acc. Chem. Res. 2003, 36, 255-263.
Since X• needs to cycle through B, C and D, these energies should be mutually
thermodynamically accessible; between any two states, there should be no
process that involves a high kinetic barrier. Now, we know what to look for in
answering the question about whether this halogenation runs with F2, Br2, or I2?
CH3CH2CH3 + X2 ® CH3CHXCH3 + HX
|
X |
ΔH (kJ /mol) |
|
F |
−463.3 |
|
Cl |
−132.9 |
|
Br |
−69.1 |
|
I |
28.1 |
|
|
|
Radical fluorination is too exothermic; it is of little use due to its lack of
selectivity. The iodination reaction is endothermic. If we could catalyze this
process and we had the products in a reaction vessel, we would isolate
reactants after the reaction completed with virtually no product remaining. So
radical iodination of alkanes is not feasible.
Using BDEs from the table above we see that the last steps in the radical chains for the Cl and Br reactions produces halogenated product exothermically.
![]()
|
X |
ΔH (kJ /mol) |
|
Cl |
−113.9 |
|
Br |
−115.3 |
|
|
|
There is one more step to consider, H atom abstraction. Here we find a thermodynamic barrier for the Br atom. As we can see below, the H atom abstraction step for Br is endothermic, but it’s exothermic for Cl. The endotherm, 46.2 kJ /mol is quite large; however, it does not have to shut down bromination. The reaction can be run with high enough internal energy so the intermediates cycle Br• quickly enough. The endothermic step makes bromination more useful by making the reaction more selective. We will discuss this in the next section.
![]()
|
X |
ΔH (kJ /mol) |
|
Cl |
-19.1 |
|
Br |
46.2 |
7.5 SELECTIVITY OF HALOGENATION
(Arthur Cammers, University of Kentucky, 2019)
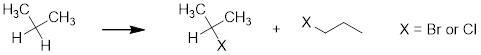
Six chemically equivalent terminal H atoms, and two equivalent internal H atoms.
There are two
sets of H atoms bound to the propane skeleton that is if we would perform one
halogenation there are two products that can result: 2-halopropane and
1-halopropane. If Mother Nature simply plays dice, we would expect a 1: 3 ratio
of 2-halopropane to 1-halopropane because there are three times as many H atoms
bound to terminal C atoms than there are H atoms bound to the internal C atoms
on the propane chain. However, we know that is not what should be observed due
to 1-propyl being stable than 2-propyl, and that these radical intermediates
are obligatory guests in the mechanistic path to products. We would expect more
2-halopropane product due to the greater stability of 2-propyl. The relatively
low energy of 2-propyl means that ΔG‡ is lower to get to
2-propyl than the ΔG‡ to get to 1-propyl. The reaction is
faster at C2 than at C1, and logically, halogenation at C2 should beat the
statistics of 1:3.
How do the
differences in the thermodynamics of Cl- versus Br- play in the product distribution
of halogenation? In particular, how does the exergonic step in the reaction
mechanism for the bromination make itself manifest in the selectivity? We might
imagine that if a reaction is not as exothermic it is likely more selective.
When you are not in a rush you have time to decide. But how do we put this into
chemistry talk? Hammond’s postulate frames the notion that less exergonic
reactions tend to be more selective in terms that we can put on the reaction
energy diagram.
Since Br is
less reactive than Cl in radical halogenation, we expect that the selectivity
for 2-bromopropane over 1-bromopropane is better than the selectivity for
2-chloropropane over 1- chloropropane.
7.5.1 Hammond’s Postulate
On the reaction
energy diagram, we chart structure and energy. It is very intuitive to think
about two chemical species as being close in stability or energy, but it is
less intuitive to realize that two features on the reaction energy diagram
could be close in structure. Proximity in structure means that two chemical
species share similar atomic parameters: bond, lengths, bond angles, and /or
dihedral angles.
Hammond’s
postulate states: The transition state between two stationary states is closer
in structure and energy to the higher-energy stationary state.
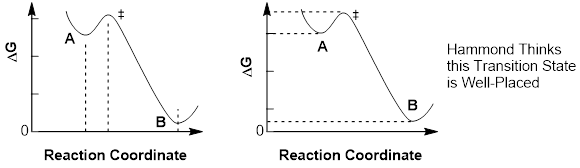
The energy diagram at the upper left shows how the transition state is closer in structure to higher-energy stationary state A than it is to lower-energy stationary state B. The energy diagram at the upper right shows how the transition state is closer in energy to higher-energy stationary state A than it is to lower-energy stationary state B.

Above, transition states placed closer in structure to the low-energy stationary state contradict Hammond’s Postulate. So, a corollary to Hammond’s notion about transition states is the following: In exothermic reactions, the transition states look like the reactant, and in endothermic reactions the transition states looks more like the product.

Why should there be any truth to Hammond’s Postulate? Consider the following nucleophilic scenario that transfers a nucleus from one reaction center to the other via a transition state.
Reactant: A─B + :C−
Transition State: (−δ) A---B----C (−δ)
Product: :A− + B─C
As the reaction proceeds from reactant to product and the ensemble increases in energy there is more bond-breaking ensuing. As we pass through the transition point the ensemble falls in energy; after this point there is more bond forming in the ensemble. Remember that decreases in energy in atomic arrays ARE the definition of bonding. Bond-breakage in the ensemble has to precede bond forming, otherwise the stationary state is no longer stationary. If there is an atomic parameter that the stationary state can change to lower its energy, it is some kind of transition state and not truly a stationary state.

Bonds break along normal vibrational modes. This means that we can separate the product-forming curve (red dash above) from the reactant-destroying curve (green dash). The potential wells are approximated above as parabolic; they basically are at the atomic distances relevant to this discussion. However, the ■ Morse potential better describes bond breakage. Of course, both curves are allowed for electrons to follow, that is a given since we start from P and R and stretch the bond.
The only other scripture that you need to attend church with Hammond is: Electrons minimize energy. If there exists an allowed state of lower energy, electrons will preferentially populate it. None of the preceding chapters, not one of them, would have been possible without accepting this statement as a fundamental property of atoms and molecules. We could not have filled out the periodic table as we did in General Chemistry, and defined ■ electron configuration for atoms (eg. For the C atom: [He] 2s2 2p2). The preference for the ground state is a foundational observable property of the electron on which chemistry is built.
Thus, above, when the reactant bond-breaking curve (green) intersects the product bond-breaking curve (red) the electrons start to make bonds along the product curve as they reverse the breaking process and form product. This same argument positions the transition state at the structural mid-point between product and reactant in an isoenergetic chemical reaction, Figure B, left. The same argument also shifts the intersection point, and thus shifts the transition state structure toward the structure of the product as the product is increased in energy, blue arrows in the Figure B, right.
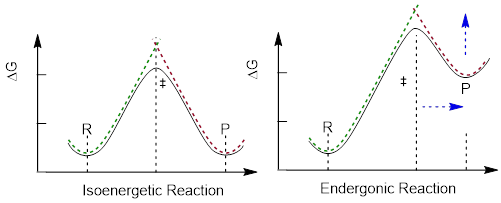
Figure B. Examples of reaction energy diagrams for an isoenergetic reaction (left) and an endergonic reaction (right).
7.5.1 Hammond’s Postulate in Selectivity of Halogenation
Hydrogen
abstraction is the elemental step that determines regiochemistry. According to
Hammond’s postulate, the transition state for hydrogen abstraction in radical
chlorination occurs earlier on the reaction coordinate than bromine’s
transition state for hydrogen abstraction.
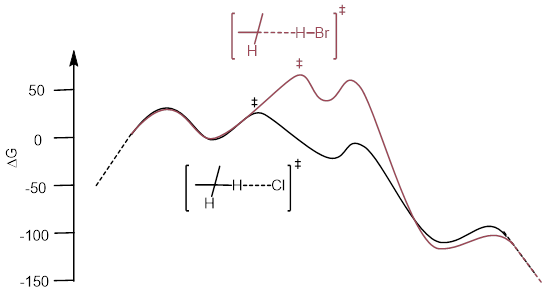
This means that the difference in energy between 1-propyl and 2-propyl are reflected in the energy of the transition more with Br than with Cl because the Br-transition state is structurally more like the radical. This must translate to a greater difference in rate between the two H atom abstractions for Br than for Cl. Giving the Br reaction as the more selective reaction. The concept is depicted in the curves below.
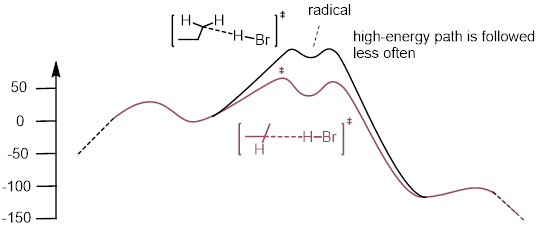
A significant difference in rate for bromine H abstraction at C1 versus C2 due to the transition state’s proximity in structure to the radical intermediate.

Not much difference in rate for Chlorine H Abstraction at C1 versus C2.
The product ratios at comparable temperatures and similar conditions look like the following: BrCH2CH2CH3 : BrCH(CH3)2 = 3:97 and ClCH2CH2CH3 : ClCH(CH3)2 = 2:3.
7.6 STEREOCHEMISTRY OF HALOGENATION
(Ashley Jolly Steelman, 2019)
What is the
stereochemical outcome for a radical halogenation reaction? When radical halogenation
produces a new chirality center or takes place at an existing chirality center,
we will see production of an equal mixture of R and S enantiomers (racemic
mixture of products). In order to explain why, it is important to examine the
radical intermediate. The radical lies in an unhybridized 2p orbital and the overall structure is considered planar.
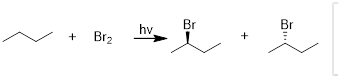

We see that
halogen abstraction can and will occur with equal probability at either face of
the plane (attacking from the top or the bottom of the intermediate) leading to
a racemic mixture of 2-bromobutane.
Exercise 7.6.2 Bromination of an alkane is usually preferred over
chlorination because of the greater selectivity. For butane, two products can
result from monobromination when treated with bromine and light however, one
product would be formed in a much higher yield than the other. (a) Draw both
monobromination products. (b) Identify which product is major versus which is
minor. (c) Draw the mechanism and demonstrate how the major product is formed.
![]()
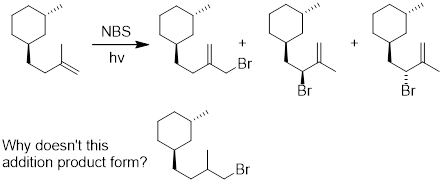
7.7 ALLYLIC / BENZYLIC BROMINATION
(Ashley Jolly Steelman, 2019)
What type of
reaction mechanism occurs when we use alkenes as our starting materials instead
of alkanes? Allylic / benzylic bromination involves the replacement of a
hydrogen atom at the allylic or the benzylic position with a bromine atom. We
can treat these two types of positions similarly, and, therefore, we will use
only the allylic position in our discussion for simplicity sake. The allylic
position is a carbon atom that is adjacent to an unsaturated carbon atom.
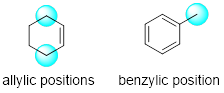
Why is the allylic C-H bond the reactive position in this reaction? The allylic C-H bonds are not your ordinary tetrahedral carbon groups. If we examine the bond dissociation enthalpies above, we see that the allylic C-H bond is the easiest to break.

However, its source of stabilization can be seen after hydrogen
abstraction at the allylic C-H bond. The
product that is formed is resonance stabilized which we know to be a great
source of stability. We do not consider the three p orbitals in the allylic
radical as separate entities. The three p orbitals form a new system of π
MOs which overall stabilize the odd electron. The p orbital is conjugated;
π bonding in MOs with electrons is maximized.

Unlike the bromination of an alkane, we cannot use Br2 to
generate our reactive radical species. If we were to dump 1 equivalent of Br2
in with an alkene we would not get our desired product. We will see later in
this class when we discuss addition reactions that alkenes react with Br2
to form vicinal dibromides (bromine adds to opposite faces of the double bond).

In order to
avoid a competition between the two reactions, scientists have discovered that
we need to use the lowest concentration of Br2 that we possibly can.
In order to do this, we use N-bromosuccinimide (NBS) as our reagent that
acts as an alternate source of a bromine radical.

Note:
We often see the
production of two products in an allylic bromination reaction. Why do you think
this is the case? Let’s examine the mechanism to answer the question.
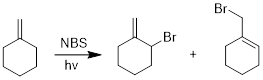
Initiation:
form reactive radical by light induced homolytic cleavage.

Propagation: (a) a bromine radical abstracts an allylic hydrogen to
produce an allyl radical. (b) the allyl radical, in turn, reacts with Br2
to form an alkyl halide and a new bromine radical.
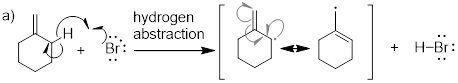


Note:
Termination is the
same for this process as was with radical halogenation. The coupling of any two
radical species will stop the reaction.
One question
that we need to address is where the Br2 came from that is necessary
for Step (b) in the propagation step of the mechanism. Once HBr is formed in
propagation (a) it can react with NBS to form Br2 in low
concentrations.

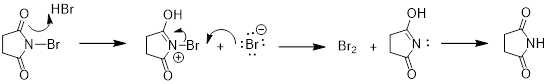
Example 7.7.1 Complete the mechanism for the benzylic bromination of methylbenzene. The overall reaction is shown. Come up with an explanation for why only one product is formed in this reaction.
![]()
Solution:
Step 1: Initiation - we need to create our reactive radical species by light-induced homolysis.
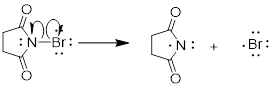
Step 2: Propagation – the reactive radical can interact with our starting materials. There are two steps in the propagation phase of the reaction.


Step 3: Termination: While we need to propagate the reaction, we also need to terminate the radical in forming our product. Note that we did that in the halogen abstraction step.
Why do we see production of only one product when A clearly has additional resonance forms? Benzene is a molecule that is termed aromatic and its aromaticity is the major contributor to why it is so unreactive. Aromatic molecules are very stable, and do not break apart easily to react with other substances. This will be a major topic of discussion in Organic Chemistry II.
7.8 ATMOSPHERIC CHEMISTRY AND THE OZONE LAYER
(Timothy Soderberg, Organic Chemistry with a Biological Emphasis II, 2016, 354-355)
The high reactivity of free radicals and the multiplicative nature of radical chain reactions can be useful in the synthesis of materials such as polyethylene plastic - but these same factors can also result in dangerous consequences in a biological or ecological context. You are probably aware of the danger posed to the earth's protective stratospheric ozone layer by the use of chlorofluorocarbons (CFCs) as refrigerants and propellants in aerosol spray cans. Freon-11, or CFCl3, is a typical CFC that was widely used until late in the 20th century. It can take months or years for a CFC molecule to drift up into the stratosphere from the surface of the earth, and of course the concentration of CFCs at this altitude is very low. Ozone, on the other hand, is continually being formed in the stratosphere. Why all the concern, then, about destruction of the ozone layer - how could such a small amount of CFCs possibly do significant damage? The problem lies in the fact that the process by which ozone is destroyed is a chain reaction, so that a single CFC molecule can initiate the destruction of many ozone molecules before a chain termination event occurs.
Although there are several different processes by which the ozone destruction process might occur, the most important is believed to be the chain reaction shown below.
1

First, a CFC molecule undergoes homolytic cleavage upon exposure to UV radiation, resulting in the formation of two radicals (step 1). The chlorine radical rapidly reacts with ozone (step 2) to form molecular oxygen and a chlorine monoxide radical. Step 3 appears to be a chain termination step, as two chlorine monoxide radicals combine. The Cl2O2 condensation product, however, is highly reactive and undergoes two successive homolytic cleavage events (steps 4 and 5) to form O2 and two chlorine radicals, which propagates the chain.
To address the problem of ozone destruction, materials chemists have developed new hydrofluorocarbon refrigerant compounds. The newer compounds contain carbon-hydrogen bonds, which are weaker than the carbon-halogen bonds in CFCs, and thus are susceptible to homolytic cleavage caused by small amounts of hydroxide radical present in the lower atmosphere:

This degradation occurs before the refrigerant molecules have a chance to drift higher up to the stratosphere where the ozone plays its important protective role. The degradation products are quite unstable and quickly degrade further into relatively harmless by-products. The hydroxide radical is sometimes referred to as an atmospheric 'detergent' due to its ability to degrade escaped refrigerants and other volatile organic pollutants.
Hydrofluorocarbons do, however, act as greenhouse gases, and are thought to contribute to climate change.
7.9 THE ORGANIC OXIDATION STATE
(Arthur Cammers, 2019)
We characterize
many chemical reactions in terms of oxidation and reduction. Formally, a
molecule is oxidized if it loses electrons or H•. Likewise, reduction is the
opposite, a gaining of electrons or H•. Gaining H− is
obviously reduction.
Earlier in the
discussion of thermodynamics in previous chapters, the conversion of simple
carbohydrates to CO2 is certainly an oxidation reaction from the
perspective of the carbon atom. In that discussion we characterized the
chemical energy in food and fuel as being at a high potential because the
electrons were caught between the C and H atoms, two atoms that are relatively
electropositive. We said that this is the way Mother Nature stores solar energy
in molecules for later release. We characterize water as not having any
chemical energy to use as fuel. In this section we are going to formalize our
concept of oxidation and reduction so we can characterize molecules and
reactions in red /ox terms.
Defining reduction and oxidation is often easier in inorganic chemistry where the oxidation state is probably more easily assessed. Organic chemists refer to the oxidation state of functional groups in a qualitative way with approximate oxidation numbers. We characterize the chemistry necessary to convert one functional group into another in terms of how these oxidation numbers change with the reaction.

You may not be immediately aware that some of the reactions above involve oxidation and others do not. This section formalizes how we need to think about reactions to know which ones involve oxidation or reduction or neither.
We can characterize oxidation states in molecules and functional groups by putting oxidation numbers on C atoms. The organic oxidation state is best thought of as an atomistic property that the chemist may have to average over a few atoms to characterize a reaction.
We define oxidation as the conversion from a functional group having a lower oxidation number to one having a higher oxidation number. We define reduction as the conversion of a functional group having a higher oxidation number to one having a lower oxidation number.
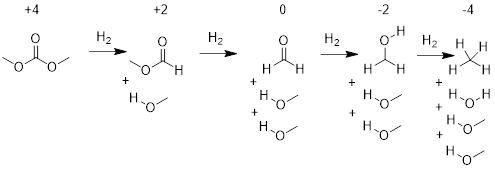
The maximum oxidation number of the C atom is +4. At the far left above in methyl carbonate we can pull groups and atoms off the central C atom of methyl carbonate to unveil the +4 oxidation number. When we pull off the atoms, we give the electrons in the bonds, at the C atoms of interest, to the most electronegative atoms. Then we count the formal charge on the C atom.

Formaldehyde is in a different oxidation state than methyl carbonate because to get from methyl carbonate to formaldehyde we have to add 2 equivalents of H2. But we can know this by performing the formalism on the structure. When we do this, we bear in mind that the H atom is electropositive relative to the C atom.
![]()
Atomistic tagging with oxidation numbers get uncomfortable when dealing with C─C bonds. We might be tempted to split the C─C bond homolytically, giving one electron each to the C atoms. However, this adds unnecessary complexity to the method. Instead we just substitute the alkyl groups with H atoms and then we apply the method.
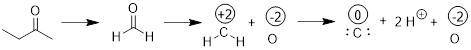
Thus, methylethylketone is in the same oxidation state as formaldehyde, oxidation state 0. Organic chemists often shortcut oxidation state nomenclature when referring to the oxidation state of functional groups: oxidation state +4 is often called the carbon dioxide oxidation state; oxidation state +2 is often referred to as the carboxylic acid oxidation state; oxidation state 0 is often the ketone oxidation state; oxidation state −2 is often called the alcohol oxidation state and oxidation state −4 is often referred to as the alkane oxidation state.
Look back at the ■ chlorination of methane. As H atom is substituted and Cl atom is added to the C atom the nominal oxidation states goes from −4 to +4. The chlorination of methane is an oxidative process and any chemistry that we would do to reverse this process, to obtain molecules associated with lower oxidation states from higher ones, we would characterize as reduction.
Exercise 7.9.1 Fred is in the lab when he notices that some solvent cans have been placed too close to an electric motor which could be the source of sparks. Someone else was not thinking, but Fred is a good citizen. There is a solvent can full of hexane and another full of carbon tetrachloride. Which one is Fred most concerned about and why?
7.9.1 Addition of Water or HX is Neither Oxidation Nor Reduction
Given the formalism to determine the organic oxidation state we can demonstrate the truth in the statement of the subheading that the addition of water or of HX is neither oxidation or reduction. Let’s do this for the addition of water across O=C in an ketone.
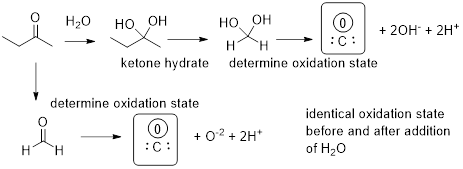
This result tells us that if we perform chemistry to change the alcohol functional group into a alkene functional group or vice versa this chemistry is in total neutral with regard to oxidation and reduction. See below to visualize how we arrived at this conclusion.
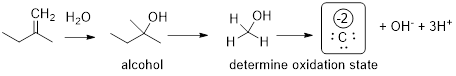
For the ketone we demonstrated that addition of H2O to the double bond did not require redox chemistry. Therefore, we can show that the alkene and the alcohol have the same oxidation state. If you can’t sort out what the oxidation state is for a double bonded functional group, add water and try to determine the oxidation state.
Since the addition of water across double bonds does not involve red /ox processes, neither does the addition of HX where X is any electronegative atom. You will see additions of HX to alkenes in the following chapters.
Exercise 7.9.2 Show that the oxidation state of the alkyne and the ketone are the same. Another way to frame this request is: Show that neither oxidation nor reduction are required to convert an alkyne into a ketone.
Hint: Step one is to add water. How many? There are different regiochemical choices to add water, what’s the best way to do so to accomplish this task?
Exercise 7.9.3 Characterize chloroform (CHCl3) as alkane, alcohol, ketone, carboxylic acid, or carbon dioxide oxidation state.
7.10 AUTOOXIDATION
(Arthur Cammers, 2019)
Autooxidation of organic compounds is the tendency to react with atmospheric oxygen to form hydroperoxides and then convert to other molecules of higher oxidation state. The process is generally slow. However, aldehydes tend to convert to carboxylic acids and thiols tend to readily convert to ■ disulfides with air oxidation relatively quickly. Sometimes this can occur overnight in an open flask. Other molecules like ethers undergo this process much slower, but autooxidation can make extended storage of ethers dangerous.
![]()

Reactions involving a radical reacting with a radical terminates the chain, but usually these products have weak σ bonds (like the O─O bond in ethers) which leads more easily to initiation. The product in the lower right is a peroxide which can be explosive. If ether ignites it will burn expansively with the ball of fire seeking oxygen in the air. Since the oxidant is included in the liquid with peroxides in the ether, ignition is much more violent. You may be familiar with the popular General Chemistry demonstration in which H2 gas is ignited followed by the ■ demonstration of the much more violent ignition of a mixture of H2 and O2 gases. The explosive nature of ether mixed with peroxides is a similar concept.

The air oxidation of aldehydes probably occurs via the aldehyde hydrate. Addition of water converts the strong sp2 CH bond into a weaker sp3 CH bond, a target for radical initialization. As in the reaction of the ether above the reaction of aldehyde with O2 leads to peroxide, but in this case, it is a peroxyacid.

The aldehyde in the presence of the peroxyacid spontaneously undergoes ■ disproportionation to two carboxylic acids.

7.11 RADICAL ADDITION OF HBr
(Arthur Cammers, 2019)
When you see
the word addition describing a reaction, the reaction of a double bond
is likely being discussed. At this point you know that HBr is a strong acid,
either from pKa arguments forwarded in this text or your knowledge from
discussions about mineral acids in General Chemistry. You are also aware that π
bonds are electron donors due to the fact that the electrons do not bond
(stabilize) as strongly as the σ bonds. The electronic availability of the
π bond tends to be somewhere between the availabilities of σ bonds
and lone pairs. It should be no surprise to you then that the HBr can add
across the CC double bond in the following heterolytic manner. Below is the
non-radical addition mechanism of HBr to the CC π bond in iso-butylene.
![]() t-butylbromide
t-butylbromide
You should also
not be surprised by the selectivity given the arguments above about the
relative stabilities of more substituted radical centers. The stabilities of
carbocations follow the same trends with the more substituted cation lower in
energy. The i-butylbromide BrCH2CH(CH3)2 addition
product is not observed. To get i-butylbromide in this addition reaction H+
would have to add to the alkene to make the primary carbocation which is a
higher-energy pathway than making the 3° cation shown above.

Organic chemists synthesize molecules from other molecules. We usually are interested in making more complex molecules from simple molecules. Isobutylene is considered a simple starting material whereas an alkyl bromide is probably more complex. It is certainly more complex from the point of view of elemental content, having one more unique element than the reactant. It would be utilitarian from the perspective of synthesis to be able to add Br to the terminus of isobutylene also. It turns out that we can do this by running a radical (homolytic) reaction instead of a heterolytic reaction. The selectivity for iso-butylbromide has the same argument in the homolytic reaction as the selectivity for tert-butylbromide does in the heterolytic reaction. Let us first consider the mechanism of radical addition and then we will discuss how we get the chemistry to switch from heterolytic to homolytic.
7.11.1 Mechanism of Radical Addition to a C-C π Bond
Given all we have discussed to this point about radical chains you know what the first step for radical addition of HBr to an alkene must be, initialization. Often this is done with peroxides. When warmed to the appropriate temperature the weak RO─OR bond breaks. The O─H bond has a high BDE so it is a powerful H atom abstractor.

This is the initialization phase of the addition reaction. In the propagation phase where reactive species cycle through spin-paired reactants and generate spin-paired products via spin-unpaired intermediates, the bromine atom enters and leaves the radical cycle in the following manner. At this stage regioselectivity for isobutylbromide is set due to the stability of the more substituted unpaired C atom.

The selectivity switches and we isolate iso-butylbromide instead of tert-butylbromide because we switch the attacking species from H+ to Br•. The behavior of the mostly p orbital at the C atom remains the same in the preference for more parallel interactions with σ bonds in the heterolytic and homolytic mechanism.

7.11.2 Reaction Conditions for Radical Addition of HBr
When the Br atom reacts with the more substituted C atom in the alkene, the reaction is called ■ Markovnikov addition. When the Br atom reacts with the less substituted C atom in the alkene the reaction is called anti-Markovnikov. You might imagine that people were puzzled about the results when good selectivity was at times observed, but mixtures were obtained in certain cases. However, once the mechanism was worked out, we know what to do to keep the results cleaner.
If we want the Markovnikov product, we avoid generating Br•. We distill the solvents to avoid species that can generate radicals. We keep our reactions away from light because hν can generate Br• from any Br2 in the mix. We also tend to run the reaction warmer to accelerate the reaction between the alkene and the acid.
If we want the anti-Markovnikov product, we do everything in our ability to generate Br• and we run the reaction at lower temperature. We heat reactions to give the reactants enough energy to collide with another molecule to make something happen. However, radical reactions have native reactivity, due to the initialization process, they are already reactive spin-unpaired molecules looking for electrons. They are already high in energy, so they tend to have lower kinetic barriers than non-radical reactions. By decreasing the temperature, we can usually slow down the heterolytic reaction without slowing down the homolytic reaction as much. The radical reaction is so much faster than the heterolytic reaction that just a small amount of initialization will promote the chain reaction. For example, it has been observed that freshly distilled 1-butene treated with HBr gives 2-bromobutane in 90% yield. However, if the reactant 1-butene has been allowed to sit in air exposed to O2, the ratios of products vary to 1-bromobutane often dominating the product mixture by as much as 3:1.


Exercise 7.11.2 Go back to the allylic bromination mechanism: Why does allylic bromination occur vs. Br• adding to the double bond in an anti-Markovnikov manner, resulting in addition across the bond?
7.12 DISSOLVING METAL REDUCTION OF ALKYNES TO ALKENES
(Susan Odom, 2019)
When we examined single electron transfer (SET) reactions in the previous section, we considered oxidative reactions. For example, when we consider the homolytic cleavage of a C-Br bond to yield two radicals, we started with having octets on both the C and Br atoms, but when the bond cleaved, each resulting radical has only 7 electrons each. In this section, we will look at the opposite scenario: Instead of electron poor species being the cause of reactions with alkenes in oxidizing conditions, here we will use reducing conditions in which reactive species produce initial radicals with too many electrons to be stable.
The reaction we will examine in this section is the dissolving metal reduction of an alkyne, as shown in the reaction scheme below. The reduction of a disubstituted – or internal – alkyne with sodium in liquid ammonia (written as Na/NH3) yields a trans alkene. While the reaction conditions are always shown as Na/NH3 without indicating the number of an equivalents, when we examine the mechanism, you will see that we need two equivalents of sodium and two equivalents of ammonia.
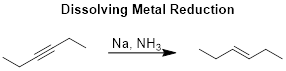
Note: Cis alkenes do not form under these reaction conditions. We will see examples of the formation of cis alkenes from alkynes when covering the catalytic hydrogenation of alkynes.
The reaction is called a dissolving metal reduction because metallic sodium is dissolved in liquid ammonia. A characteristic of this reaction is its brilliant blue color. Given that the boiling point of ammonia is -33 °C, without providing temperature information under the reaction arrow, you know this must be a cold reaction given the liquid state of ammonia. For practical reasons – in that dry ice / acetone baths are used to cool the reaction flask – these reactions are usually conducted at -78 °C. You may see similar reaction conditions, including Li instead of Na or the solvent tetrahydrofuran (THF) instead of ammonia.
When an alkyne is introduced into the sodium/ammonia solution, it accepts a single electron into the π* LUMO, forming a radical anion. By definition, a radical anion is a species that is negatively charged and has an odd number of electrons.

The reason for the formation of the trans-alkene instead of the cis-alkene is due to differences in stability between the two radical anions that could form in the first step of the reaction mechanism. When adding the solvated electron to the alkyne, either the trans or cis radical anions can form. The trans radical anion is lower in energy due to the alkyl groups being farther from each other. In the cis version, steric repulsion of the ethyl groups leads to a higher energy conformation by about 8 kJ /mol. Thus, it is the trans radical anion that exists at this stage and leads to the trans alkene as the eventual product of this reaction mechanism.
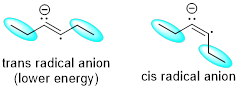
In the mechanism above, the last protonation of the anion can happen as indicated, but some alkene products are not basic enough to take the proton off NH3. These sit in solution as anions until the chemist quenches the reaction with a proton source, usually the controlled addition of an alcohol like iPrOH then water. The pKa of propene is 43 whereas the pKa of NH3 is 38. If we are reducing propyne to propene by this method, we would expect that propene (not its conjugate base) be the product in the mechanism above.
Exercise 7.12.1 Dissolving metal reductions are conducted with an excess of sodium and ammonia, the reduction only proceeds from the alkyne to the alkene, not further to form an alkane. Provide an explanation for why the second reduction does not occur. (Hint: think about the relative stability of intermediates in the first vs second reductions.)
You now know enough types of reactions to complete a problem in multi-step reaction sequence – meaning that two or more reactions are involved in a linear progression. Try your hand at predicting the major product(s) below.
Exercise 7.12.2 Predict the major product or products of the reaction sequence below.
![]()
NBS = 
7.13 BIRCH REDUCTION
(Susan Odom, 2019)
Dissolving metal reductions can also be accomplished in other pi systems. An example of this is the Birch reduction, in which a dissolved metal in a protic solvent can be used to reduce benzene rings. An overview of the reaction is shown below.
![]()
Let’s take a look at the mechanism. In this example, we will use Na/NH3 as our reagents. In the first step, sodium donates an electron to benzene to make a radical anion. The basic anion is protonated with ammonia, affording a neutral radical. This radical is reduced by another equivalent of sodium, producing an anion. In a final step, the anion is protonated by another equivalent of ammonia, forming the cyclohexadiene derivative shown below.


Why did 1,4-cyclohexadiene form rather than its 1,3-isomer? First, let’s consider delocalization of our intermediate species. Although each intermediate is drawn with one bond-line structure, you must appreciate that the lone pair and radical are delocalized in the system. Focusing on the second carbanion that forms, we have three good resonance structures, and it is possible for three sites to be protonated, yet one of them dominates.

Based on what we know about the relative weighting of resonance structure, we would characterize A, B, and C as equally weighted. Further one might expect that because A and C are equivalent structures, you may have double the odds of protonating the positions next to the sp3-hybridized carbon atom. Yet the product that dominates is 1,4-cyclohexadiene, and this pi bond arrangement is common over multiple benzene derivatives, whether the benzene ring contains electron-donating or withdrawing substituents. Often multiple products result.

One explanation for the favoring of 1,4-cyclohexadiene was proposed by Hine who described the principle of least motion. Essentially, Hine’s explanation says, “…elementary reactions will be favored that involve the least change in atomic position and electronic configuration.” [Reference below] If one assigns bond order to the radical anion based on the resonance structures A, B, and C above, there are 2 bonds that are present 2/3 of the time and two bonds that are present 1/3 of the time, giving an approximate bond order as shown below. When converting the anion to the product, there is less change when leaving the pi bonds opposite one another.

If the 1,4-diene is formed, the mathematical changes can be summarized as 0 + 0 + 1/3 + 1/3 + 1/3 + 1/3 = 1 whereas forming the 1,3-diene would be 0 + 0 + 2/3 + 2/3 + 2/3 + 2/3 = 1 1/3. The principle of least motion should be intuitively connected to this kinetic product. If the 1,3- and 1,4-diene were to equilibrate, the reaction would produce the more stable, conjugated 1,3-diene.
This explanation is the simplest we can provide using bond-line structures. In fact, we would require molecular orbital theory to give a better answer, and if you are interested in a more advanced perspective, we recommend that you go to office hours to discuss further.
Exercise 7.13.1 Why does this reaction stop at the diene rather than proceed to cyclohexane or cyclohexene?
Exercise 7.13.2 Two possible products can result from the reduction of benzoic acid when treated with Na/NH3/EtOH. Draw both products.

References:
Original source on principle of least motion: Hine, J. J. Org. Chem. 1996, 31, 1236.
This content is summarized in greater detail in section 15-14 of “March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 5th Edition,” by Michael B. Smith and Jerry March.
7.14 RADICAL POLYMERIZATION
(Susan Odom, 2019)
Polymerization reactions occur when monomer units react with each other to form a chain containing the monomer units. You encounter polymerization reactions in your day to day life. For example, if you get your gel nails at the nail salon, the hardening process is a light-induced polymerization of methacrylate – a process called photocuring. This is the same process used to harden composite tooth fillings; here the monomer is methacrylic acid. You also encounter polymers in every plastic product you use – from your water bottle to your grocery bag to your car’s bumper. These polymers are made by one of two general routes: chain growth or step grown.
In chain growth reactions, a polymer is grown from a single activated unit, with each additional unit adding to the active end of a growing polymer chain. These are often the polymers that are named poly-something (e.g. polyethylene, polypropylene, polystyrene). In step growth polymerizations, two types of monomer units combine and a small molecule (e.g. water) is removed. Polymers like nylon – the elastomeric component of stockings and tights – are made this way.
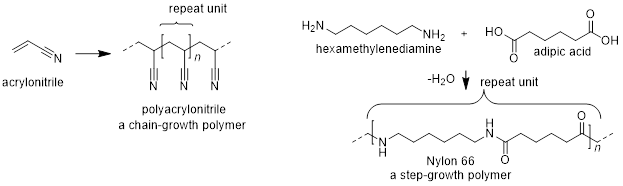
Polymer chemistry is its own field, and the synthesis alone would take a full semester to cover. Here we will take a quick glance at radical polymerization reactions – a type of chain-growth polymerization – so you are familiar with the basic characteristics and requirements.
7.14.1 Overview of a Radical Chain-Growth Polymerization Reaction
Radical polymerization reactions have three mechanistic steps: initiation, propagation, and termination. In initiation, an input of energy results in the activation of a radical initiator – the site from which the polymer chain will grow. In propagation, the activated radical initiator reacts with a monomer unit, and continues to react with additional monomers to build the polymer chain. Lastly, in termination, the radical species at the end of the growing polymer chain reacts to deactivate the propagating species, ending the growth of the polymer chain. The next sections will briefly describe each step.
7.14.2 Initiation
The first step of a polymerization reaction is to input energy to activate a radical initiator. The desired outcome is the homolytic cleavage of a bond to form two radical species. Common ways activate initiators include (i) applying heat in a thermal decomposition reaction and (ii) irradiating a sample with light – often in the UV range – in a photolysis reaction. Peroxides such as benzoyl peroxide (BPO) are common thermal initiators, and azo compounds such as azoisobutyrylnitrile (AIBN) are initiated photolytically. Both mechanisms are shown below.
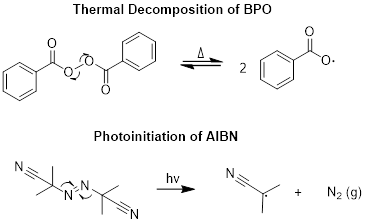
Notice that in the BPO initiator, a weak O-O bond is present. We need at least one of the following characteristics in a good initiator: a weak bond, a stabilize radical product, or an irreversible reaction.
Exercise 7.14.1 Why are the activation of AIBN and the second step in the activation of BPO shown as forward arrows? In other words, why are these reactions irreversible?
7.14.3 Propagation
After an initiator is activated, it reacts with a monomer unit to form a propagating species. The radical site in this species is called the propagating site. The propagating site reacts with another monomer unit, moving the propagating site to the newly adopted monomer. This process continues until monomer is consumed or until the propagating radical encounters another species that leads to its deactivation.

A variety of monomer units can be used in radical polymerization reactions. These are usually alkenes, although they may contain additional functional groups that are incorporated to impart certain physical properties into the resultant polymer such as toughness, elasticity, solubility, and adhesion. Some examples of monomer units are shown below.

Figure C. Common monomers used in controlled radical polymerizations.
Exercise 7.14.2 Draw the repeat unit for the chain-growth polymer that results from chain-growth radical polymerization using the monomers in Figure C. For example, with ethylene, your answer would be the following:
![]()
Exercise 7.14.3 Imagine you conducted a polymerization with 1 mmol of AIBN initiator and 100 mmol of the monomer propylene (CH2=CHCH3). What is the average number of propylene units each polymer chain would contain?
7.14.4 Termination
The last step of a chain-growth polymerization is termination. In this step, the propagating site reacts to deactivate the radical. Numerous reactions can lead to termination. One is the termination of two propagating sites by combining with each other. When two propagating species encounter each other, another possibility for termination is disproportionation; here one propagating chain abstracts a H atom from another propagating chain, leaving the second chain to form an alkene – as a diradical would be too high in energy in comparison.
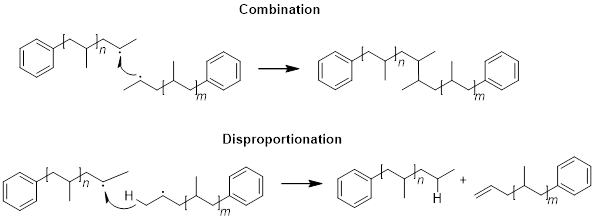
The longer polymer chains become, the less likely this mechanism is to occur as the likelihood of two propagating sites encountering each other decreases.
In another radical-with-radical deactivation, the propagating site could encounter an activated initiator that did not start propagating, and these two species could recombine.

Radical polymerization reactions are susceptible to reaction with oxygen gas, which is why most of these polymerizations are conducted under nitrogen or argon gas with rigorous steps taken to exclude oxygen. Shown below is a termination mechanism in which the propagating radical reactions with oxygen, producing a less reactive propagating species.

Oxygen is one of many polymerization reaction inhibitors. In many cases, inhibitors are added to bottles of monomers to prevent polymerizations from automatically starting in the shipping container, rendering the monomer an unusable solid block.
7.14.5 Kinetics
Three equations need to be used to describe the kinetics of a polymerization, because each step – initiation, propagation, and termination – is dependent on a different combination of species.
Look at the rate equations below and see if you can make sense of them. The term f is the efficiency of the initiator; kd, kp, and kt are the constants for initiator dissociation, chain propagation, and termination, respectively. [I], [M], and [M·] are the concentrations of initiator, monomer, and propagating species, respectively.
Initiation rated
= 2kdf [I]
Propagation ratep = kp[M][M·]
Termination ratet
= 2kt[M·]2
In the initiation step, the initiator is the only species on which concentration is dependent. The efficiency of the initiator (f) depends on bond strengths and stability of the resultant radicals. In photoinitiated reaction, the wavelengths at which the initiators absorb light vs the wavelength of the light source as well as the molar absorptivity at those wavelengths. Propagation requires reaction of the propagating chain with monomer, which is why both terms are included. Lastly, if two propagating chains react – and are the only mode of termination – then the reaction requires that two chains encounter one another.
KEY TO CHAPTER 7 EXERCISES
(Arthur Cammers, Ashley Steelman, and Susan Odom, 2019)
Exercise 7.1.1 Answer: Just like •H would make H2, these
radicals would dimerize.
a) •CH3 +•CH3 ─► H3C─CH3

Exercise 7.2.1 Put the 2-propyl radical in the appropriate conformation for hyperconjugative interaction. Show this interaction in a ■ Newman Projection.
Exercise 7.2.1 Answer:
***insert answer***
Exercise 7.2.2 Answer:

When you compare the bond breaking reactions, on the product side you see •H and the corresponding radical so the difference must be BDE. In kJ /mol: 4) ~410, 3) ~397, 2) ~381, 1) ~356; compare these numbers to methyl C‑H BDE of 435 kJ /mol.
Exercise 7.4.1 Answer: How would you proceed with this exercise in an
effective manner? First write out the equations and make sure the mass is
balanced.

There are three intermediates energies and two halogens to consider.
|
X |
B |
C |
D |
|
Cl |
|
−19.1 |
−113.9 |
|
Br |
|
46.2 |
−115.3 |

We can better visualize the differences in the two reactions if we put them on the same reaction energy diagram. Above the Br path is labelled with ‘. The concentration of the radical species is small even after initialization. At C’ equilibrium acutely decreases the concentration of radical species. At C, equilibrium enhances the concentration of radical species.
Exercise 7.6.1 Provide a reaction mechanism for the reaction below.
![]()
Exercise 7.6.1 Answer:
***insert answer***
Exercise
7.6.2 Bromination
of an alkane is usually preferred over chlorination because of the greater
selectivity. For butane, two products can result from monobromination when
treated with bromine and light however, one product would be formed in a much higher
yield than the other. (a) Draw both monobromination products. (b) Identify
which product is major versus which is minor. (c) Draw the mechanism and
demonstrate how the major product is formed.
![]()
Exercise 7.6.2 Answer:
***insert answer***
Exercise 7.9.1 Answer: Due to the oxidation state of CCl4, we are not concerned at all with it catching fire. In fact, carbon tetrachloride can be used to extinguish flames, and would be, if it not for the fact that a lot of harmful vapors would be in the air because of its low boiling point. Hexane, on the other hand, is highly flammable due to its potential to rapidly, exothermically oxidize.
Exercise 7.9.2 Answer: Add two water molecules to a plain vanilla alkyne and add the O atoms to the same C atoms. At this point remove one water to get to the ketone, or determine the oxidation state of the 1,1-alkyne hydrate.

Exercise 7.9.3 Answer: +2, or the same as a carboxylic acid
Exercise 7.11.1 Answer:
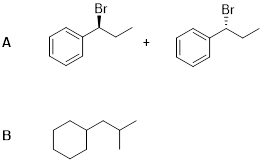
Exercise 7.11.2 Answer: Br• does add to the double. This reaction is fast, which we know from the discussion of radical addition of HBr. However, in the NBS reactions, there is no HBr present to donate H•, so the addition reverses.
Exercise 7.12.1 Answer: The C atom in the sp2 hybridized C in the first radical cation (en route to the alkene) is more electronegative than the sp3 hybridized C atom in the second radical cation (en route to the alkane). The lesser stability of anion on an sp3 hybridized C atom is a sufficient barrier to prevent the second reduction from occurring.
Exercise 7.12.2 Answer:
![]() (via trans-2-butene,
which results from the first step)
(via trans-2-butene,
which results from the first step)
Exercise 7.13.1 Answer: The conditions for the Birch
reduction are not sufficiently reducing to transform an isolated double bond
into a single bond. Like with the dissolving metal reduction in which one pi
bond is reduced per alkyne, here reduction of one bond per benzene ring occurs.
Exercise 7.13.2 Answer:

Exercise 7.14.1 Answer: Because the product of AIBN formation (N2) and BPO decomposition (CO2) gases that leave the reaction mixture, they are unavailable to react with the radical and reverse to the starting components.
Exercise 7.14.2 Answer:


Exercise 7.14.3 Answer: 50. Each mmol of AIBN produces 2 mmol of initiator. Therefore, the ratio of initiator to monomer is 2:100, or 1:50.]
Exercise 7.14.4 Answer: (a) two propagating chains meeting, (b) ratet = kt[M·][O2]
CHAPTER 7 PRACTICE PROBLEMS
(Arthur Cammers, Ashley Steelman, and Susan Odom, 2019)
PP 7-1 In each set of molecules determine which radical is the most stable.

PP 7-2 Draw all resonance structures of the radical below using fishhook arrows.
![]()
PP 7-3 Which
CH bond is the weakest in the molecule below.
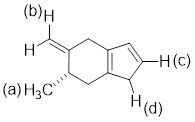
PP 7-4 Determine the reagent(s) necessary to perform the following reactions.
![]()

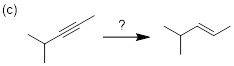
![]()
PP 7-5 Halogenation of alkanes with elemental bromine is usually preferred over chlorination because of the greater selectivity of bromination. For 3-methylpentane, three products can result from monobromination when treated with bromine and light. (a) Draw ALL monobromination products (disregard stereochemistry). (b) Identify which product is major versus which are minor.
![]()
PP 7-6 Determine the major product of each of the following reactions.
…

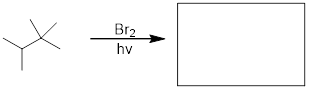

PP 7-7 A free radical bromination was performed on the chiral starting material below (stereochemistry shown). What is the stereochemistry of the major product (not shown)?
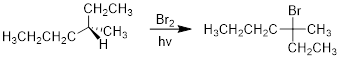
PP 7-8 Propose a mechanism for the following transformation.
![]()
PP 7-9
Mechanism:
Provide a step-by-step mechanism for the reaction below showing how the product
is formed. Show each intermediate that is formed including any formal charges.
Use the appropriate arrows to show the electron flow for each step.

PP 7-10 Read this experimental section from Tuleen, D. L.; Hess, B.A. J. Chem. Ed. 1971, 48, 477. Experimental Section: To a mixture of p-toluic acid (2.72 g, 0.02 mole, Aldrich) and N-bromosuccinimide (3.6 g, 0.0202 mole, Aldrich) was added benzoyl peroxide (0.2 g) in such a manner that none of it remained on the ground joint. Carbon tetrachloride (25 mL) was added and the mixture was refluxed for 1 hr with occasional swirling. The contents of the flask were chilled in an ice bath, then filtered with suction. The resultant solid was washed with three 10 mL portions of pentane, pressed dry, and transferred to a beaker. Water (50 mL) was added and the resultant slurry was stirred briefly to dissolve succinimide. The crude product was separated by suction filtration and allowed to dry.
(a) Why is CCl4 a good solvent for this reaction?
(b) What were they trying to make starting with ■ p-toluic acid?
(c) What did the Tuleen and Hess make given the NMR spectrum of the product? The IR of the product indicates the presence of the carboxylic acid.
(d) Can you draw a mechanism for this reaction without looking back at any material?

PP 7-11 Draw a reaction mechanism for the polymerization of vinyl chloride to form poly(vinyl chloride) (PVC), –(CH2CHCl)n–, beginning with the photoinitiation of benzoyl peroxide (BPO). Label the initiation, propagation, and termination steps. (The type of termination reaction you use is your choice.)
PP 7-12 The polymer poly(vinylidenedifluoride) (PVDF), –(CH2CF2)n–, is used electronics, biomedical applications, and high-temperature processes. This polymer is made in a free radical process. (a) What one monomer is used in the synthesis of PVDF? (b) It is conceivable that PDVF could be prepared from a combination of two monomers – assembled in an alternating arrangement in the polymer chain – what would these two monomers be?
PP 7-13 Chain growth radical polymerization reactions are usually exothermic processes. In practice, radical polymerizations are often conducted at low temperatures, as higher temperatures favor depolymerization. Provide an explanation for this phenomenon – that higher temperatures favor depolymerization. (Hint: Think about what you learned about thermodynamics in earlier chapters)
PP 7-14 Cyclic monomers can be used in chain growth radical polymerization reactions. In some cases, these rings are opened during the reaction. For each of the polymerization reactions shown, draw a mechanism showing the ring-opening propagation step. (Note: these mechanisms are difficult compared to the others you’ve practiced! Try not to be discouraged.)

KEY TO CHAPTER 7 PRACTICE PROBLEMS
PP 7-1 Answer:

PP 7-2 Answer

PP 7-3 Answer: The proton labeled (d) is the weakest
proton and is the bond that is easier to break. Upon removal of the proton the
lone pair of electrons will be stabilized by resonance.
PP 7-4 Answer:
![]()
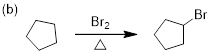

![]()
PP 7-5 Answer:

PP 7-6 Answer:
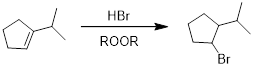

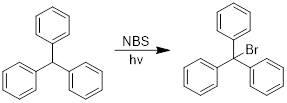
PP 7-7 Answer:
PP 7-8 Answer:
PP 7-9 Answer:
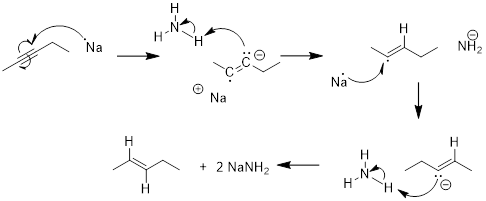
PP 7-10 Answer:
(a) CH2=CF2
(b) CH2=CH2 and CF2=CF2
PP 7-11 Answer: Recall the equation for the Gibbs free energy of a reaction, ΔG = ΔH – TΔS. At low temperatures the TΔS term has little impact on ΔG of the reaction. As the temperature increases, the entropy term is given more weight. Because depolymerization is entropically favorable, the polymerization becomes less favorable as the TΔS term grows larger, thus the reason to limit the temperature of polymerization reactions.
PP 7-12 Answer: See Schemes 1 and 2 here.
PP 7-13 Answer: ΔG = ΔH −TΔS. Many pieces increases the ability of the substance to absorb heat. The monomer state of the system is entropically favored over the polymer. As the polymer acquires heat the system will favor the monomer in accordance with ΔG = ΔH −TΔS with −TΔS contributing negatively to ΔG.
PP 7-14 Answer:
